Introduction
Having established the scientific foundation in the early stages of dissolution method development (Steps 1–6) in the previous article: Dissolution test Method development part 1, the next phase shifts toward method optimization, validation, and formalization. At this point, the preliminary experiments have provided a clear understanding of the drug’s solubility, appropriate apparatus selection, and discriminatory media conditions. However, these initial trials alone are not sufficient for regulatory acceptance or routine quality control use.
The upcoming steps (7–12) are aimed at ensuring the method is not only scientifically justified but also robust, reproducible, and defensible for its intended purpose — whether that is routine QC release, stability studies, generic bioequivalence, or IVIVC modeling. This involves refining the method to the mildest yet discriminatory conditions, validating its performance characteristics per international guidelines, defining acceptance criteria, and documenting the method for regulatory submission.
In essence
Part 2 transforms a “working method” into a validated, regulatory-ready analytical procedure that withstands the scrutiny of both internal QA systems and global health authorities.
Step 7: Optimize method for intended purpose
At this stage, the preliminary screening studies have identified feasible apparatus, medium, and operating conditions. The focus now shifts to refining the method so that it is both discriminatory and practical for its intended use.
- Discriminatory Power: The dissolution method should be able to detect meaningful changes in product quality attributes, such as variations in tablet hardness, particle/granule size distribution, compression force, or coating level. Methods that fail to differentiate between acceptable and unacceptable formulations are not suitable for QC or regulatory purposes.
- Mildest Suitable Conditions: Conditions should not be excessively harsh (e.g., unnecessarily high agitation rates, elevated surfactant concentrations, or extreme pH adjustments) as these may mask differences between formulations. Conversely, conditions that are too mild may result in incomplete release or high variability.
- Balancing Clinical Relevance and QC Feasibility: Where possible, selected conditions should reflect biorelevant environments (e.g., pH 1.2, 4.5, 6.8 media) while maintaining the reproducibility and simplicity required for routine QC laboratories.
- Regulatory Guidance: Reference USP <1092> and FDA guidance documents emphasize that the final method should reliably reflect formulation/process changes and be justified with experimental data.

Outcome
The optimized method becomes the candidate final method that can proceed into validation. It provides consistent dissolution profiles, demonstrates discriminatory ability, and meets the product’s Quality Target Product Profile (QTPP) requirements.
Step 8:Validation & system suitability
Once the method is optimized, it must be validated in compliance with ICH Q2(R2) requirements. Validation should confirm that the method is reliable, reproducible, and fit for its intended use.
- Specificity: Demonstrate that the method measures only the analyte without interference from excipients, degradation products, or dissolution media components.
- Linearity: Establish the calibration curve across the working range (typically (Q-45%)–120% of the expected concentration), with correlation coefficient (R²) ≥ 0.99 .
- Accuracy: Spike recovery studies at multiple levels (e.g., (Q-45%)%, 100%, 120%). Watch the video below for more information about the range of analytical procedure of dissolution.
- Precision: Repeatability (intra-day) and intermediate precision (inter-day, analyst-to-analyst). %RSD should be within defined limits (generally ≤ 2%).
- Filter compatibility: Verify that filters do not adsorb analyte and do not cause particle shedding; compare filtered vs centrifuged samples.
- Solution stability: Confirm analyte stability in the medium over the intended sampling period.
- System suitability testing (SST): Define acceptance limits for parameters such as plate count, tailing factor, and precision of replicate injections. SST should be run before each sequence to confirm the system is performing properly.
Outcome
Successful validation ensures that the dissolution method is not only scientifically justified but also regulatory-compliant and reproducible across laboratories. The validated method can then be applied to QC, stability testing, and regulatory submissions.
Step 9:Acceptance criteria & sampling plan (Q, S1/S2/S3 or L1/L2/L3)
Following validation, the method must be linked to clear acceptance criteria that define how dissolution data are interpreted for batch release and regulatory compliance. These criteria ensure consistency and provide a statistically sound basis for decision-making.
a) Immediate-release (IR) products:
- Acceptance is typically based on the USP staged testing scheme:
- Stage 1 (S1): 6 units tested; each unit must be ≥ Q + 5%.
- Stage 2 (S2): If S1 fails, test an additional 6 units (total 12). The average of all 12 must be ≥ Q, with no unit < Q − 15%.
- Stage 3 (S3): If S2 fails, test an additional 12 units (total 24). The average of all 24 must be ≥ Q, with no unit < Q − 25%.
- The Q value (percentage dissolved at the specified time) should be defined based on clinical relevance, reference product performance (for generics), or compendial standards.
b) Modified-release (MR) products
- Multi-time-point specifications are established (e.g., 20–30% at 1 h, 40–60% at 4 h, NLT 80% at 12 h).
- Evaluation uses L1/L2/L3 criteria:
- L1: Each of 6 units lies within the acceptance limits.
- L2: If L1 fails, 6 more units tested (total 12). The average of all must remain within acceptance limits, with tighter bounds on individual units.
- L3: Final stage (24 units), ensuring population-level compliance.

Justification of criteria:
- Acceptance limits must be scientifically justified, taking into account:
- Clinical performance requirements (IVIVC/PK correlation, BE targets).
- Innovator/reference product profile (for generics).
- Compendial monographs (where available).
- Historical data on manufacturing variability.
Outcome
The defined acceptance criteria and staged sampling plan provide a statistically robust decision framework that ensures product consistency, supports regulatory approval, and aligns with pharmacopeial requirements.
Step 10: Robustness & ruggedness testing
Even a well-validated dissolution method must demonstrate resilience to normal, expected variability in laboratory conditions. Robustness and ruggedness studies confirm that the method consistently produces reliable results under such conditions.
Robustness:
- Conducted by introducing small, deliberate variations in method parameters to assess their impact on dissolution results.
- Typical variables include:
- pH of medium: ± 0.05 units
- Agitation speed (rpm): ± 5 rpm
- Temperature: ± 0.5 °C
- Medium volume: ± 50 mL
- Sampling time: ± 2 minutes
- Results should demonstrate that such changes do not significantly affect the dissolution profile or acceptance outcome. If results are highly sensitive, the parameter should be more tightly controlled in the SOP.
Ruggedness:
- Evaluates reproducibility of the method under different operational conditions, typically involving:
- Different analysts
- Different dissolution apparatus or HPLC/UV instruments
- Different laboratories (in the case of method transfer)
- Ruggedness ensures the method is transferable and not dependent on a single operator or equipment set.
Outcome
Demonstrating robustness and ruggedness ensures the dissolution method is practical, reliable, and transferable, supporting its routine application in global QC and stability programs.
Step 11: Troubleshooting decision trees
Despite thorough optimization and validation, dissolution methods may occasionally yield unexpected or out-of-trend results. Establishing structured troubleshooting pathways helps laboratories systematically identify and correct issues without unnecessary repetition of full-scale studies.
Common issues and root causes:
- Low or incomplete release
- Incorrect medium composition or preparation (e.g., pH drift, missing surfactant).
- Inadequate deaeration leading to air bubbles on dosage form.
- Mechanical issues such as basket clogging or paddle height misalignment.
- High variability between units
- Poor content uniformity or hardness variability in the dosage units.
- Inconsistent sampling (timing or volume errors).
- Improper degassing of medium causing erratic hydrodynamics.
- Unexpected profile changes
- Product instability (e.g., moisture uptake, polymorphic transformation).
- Manufacturing process shifts (e.g., granulation size, coating changes).
- Incorrect medium preparation (e.g., buffer strength deviation).
- Analytical problems (HPLC/UV assay)
- Increasing backpressure or poor peak shape (clogged guard column, contaminated mobile phase).
- Interference from degradation products or excipients.
- Inadequate filter compatibility assessment.
Decision tree approach:
- Begin with mechanical/system checks (apparatus alignment, paddle wobble, deaeration).
- Verify medium preparation (pH, ionic strength, surfactant concentration).
- Confirm sampling and assay integrity (filters, dilutions, stability).
- Investigate product factors (tablet hardness, coating thickness, storage conditions).
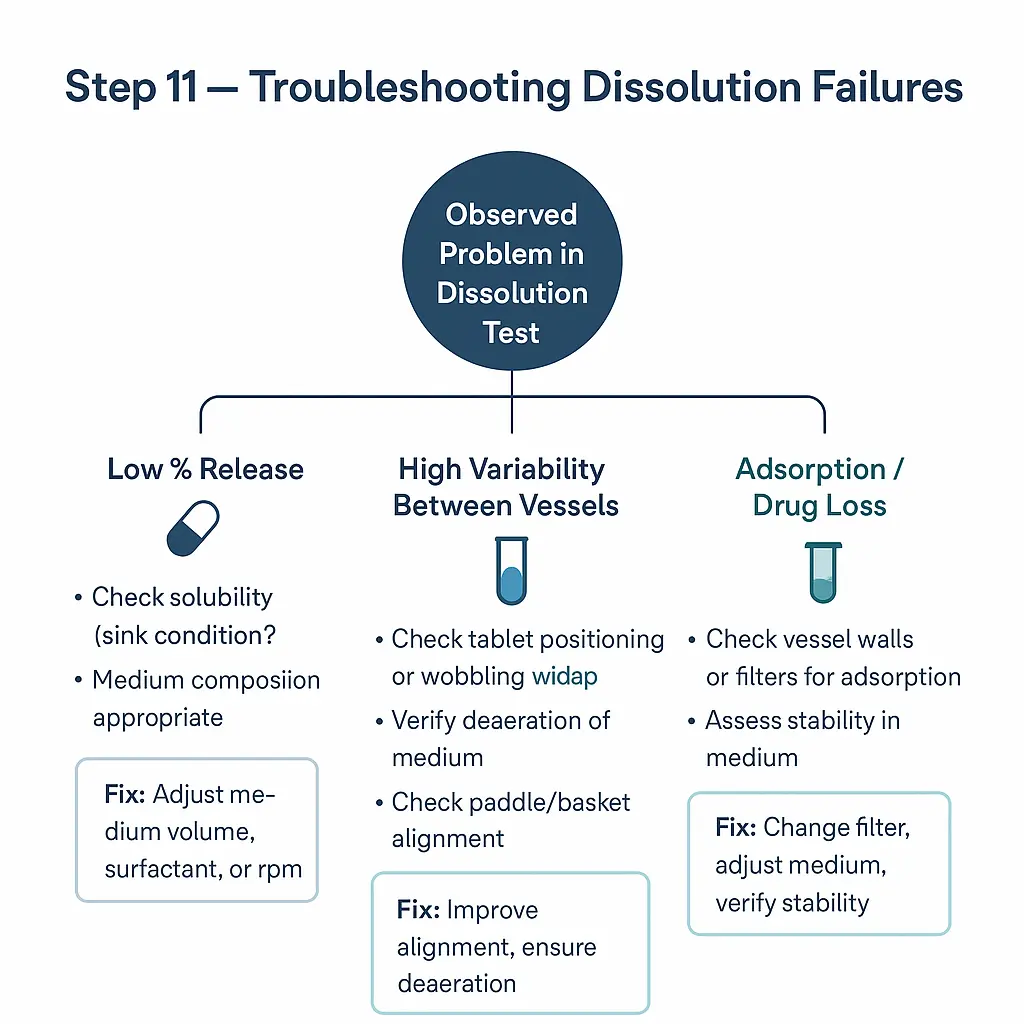
Outcome
Troubleshooting decision trees allow efficient root cause identification and corrective action, minimizing downtime and avoiding unnecessary repeat testing. Well-documented troubleshooting protocols also support regulatory inspections and enhance laboratory quality culture.
Step 12: Documentation & regulatory submission notes
The final step in dissolution method development is to ensure that the method is fully documented, justified, and submission-ready. Regulatory authorities expect a clear demonstration that the method is scientifically sound, reproducible, and directly linked to the product’s QTPP and CQAs.
Documentation requirements include:
- Method description: Apparatus, medium composition, agitation speed, temperature, sampling plan, filtration, and analytical assay.
- Scientific justification: Rationale for each parameter, including medium selection (pH relevance, sink condition data, surfactant justification) and apparatus choice.
- Supporting data:
- Solubility data across physiological pH range.
- Sink condition assessment.
- Filter compatibility and solution stability results.
- Robustness and ruggedness evaluations.
- Validation data per ICH Q2(R1).
- Comparison to compendial or reference methods: If a compendial method exists, explain alignment or justify deviations. For generics, provide evidence of profile similarity to the reference product.
- System suitability requirements: Explicit SST criteria to ensure ongoing method performance.
Regulatory context:
- In the CMC section of regulatory submissions, dissolution methods must be presented with complete justification.
- Agencies such as the FDA, EMA, and WHO expect transparent rationale for any non-standard choices (e.g., use of surfactants, reduced volumes, or atypical apparatus).
- Proper documentation facilitates method transfer to QC laboratories and supports global regulatory acceptance.
Outcome
Thorough documentation transforms the dissolution method from a laboratory procedure into a regulatory-compliant analytical standard, ensuring it withstands scrutiny from health authorities and remains reliable throughout the product lifecycle.
Conclusion:
Dissolution method development is a systematic, stepwise process that evolves from initial exploratory studies into a validated, regulatory-accepted analytical tool. While the early stages (Steps 1–6) focus on understanding the physicochemical properties of the drug and identifying suitable apparatus, media, and preliminary conditions, the later stages (Steps 7–12) emphasize refinement, validation, and formalization.
Through careful optimization, validation per ICH Q2(R1), definition of acceptance criteria, robustness/ruggedness testing, and structured troubleshooting pathways, the method becomes a reliable quality standard. Proper documentation and justification, aligned with USP, FDA, EMA, and WHO guidance, ensure that the method not only supports internal quality systems but also meets global regulatory expectations.
Ultimately, a well-developed dissolution method serves as more than just a QC test it is a critical link between product design, clinical performance, and regulatory assurance, enabling consistent product quality throughout the lifecycle of the drug.