Introduction
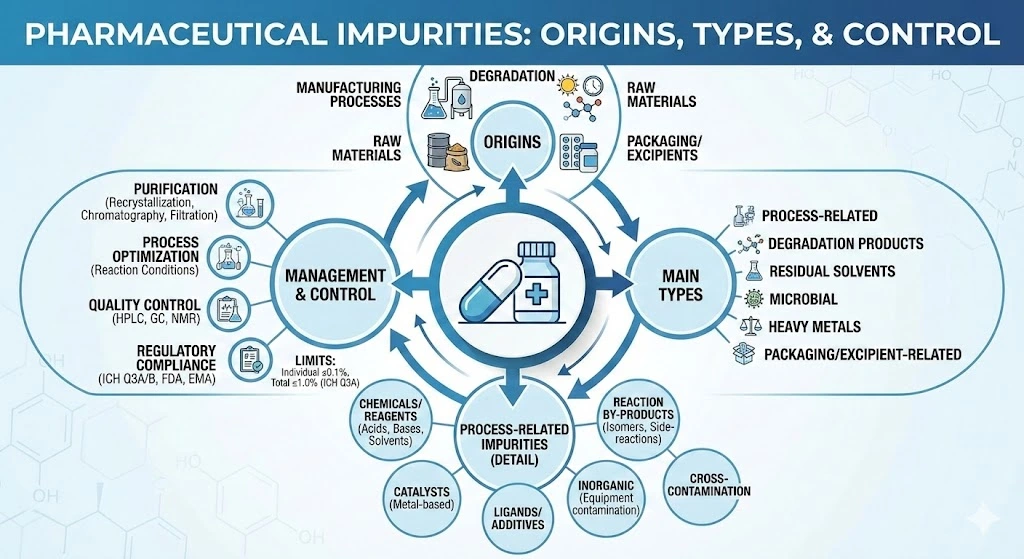
The definition of Impurities in drug products is that they are any substances present in the final pharmaceutical formula that are not the intended Active Pharmaceutical ingredient (API). They can arise from various sources, including the manufacturing process, degradation over time, and even the raw materials used in production. The presence of those impurities undoubtedly poses a risk to the patient’s safety, can affect the drug’s performance, efficacy, and may also interfere with regulatory approval.
Impurity Types
- Process–Related Impurities: These are defined as by-products, side-products, or unreacted starting materials formed during the synthesis or purification of the API. Catalysts, reagents, solvents, metal traces, etc, are considered process-related impurities that may be introduced during manufacturing.
- Degradation Products: The drug substance or product may be exposed to various environmental conditions such as light, heat, or moisture. These conditions can lead to the formation of degradation products, which may reduce therapeutic activity and potentially compromise patient safety. Drug Degradation Mechanism | Veeprho
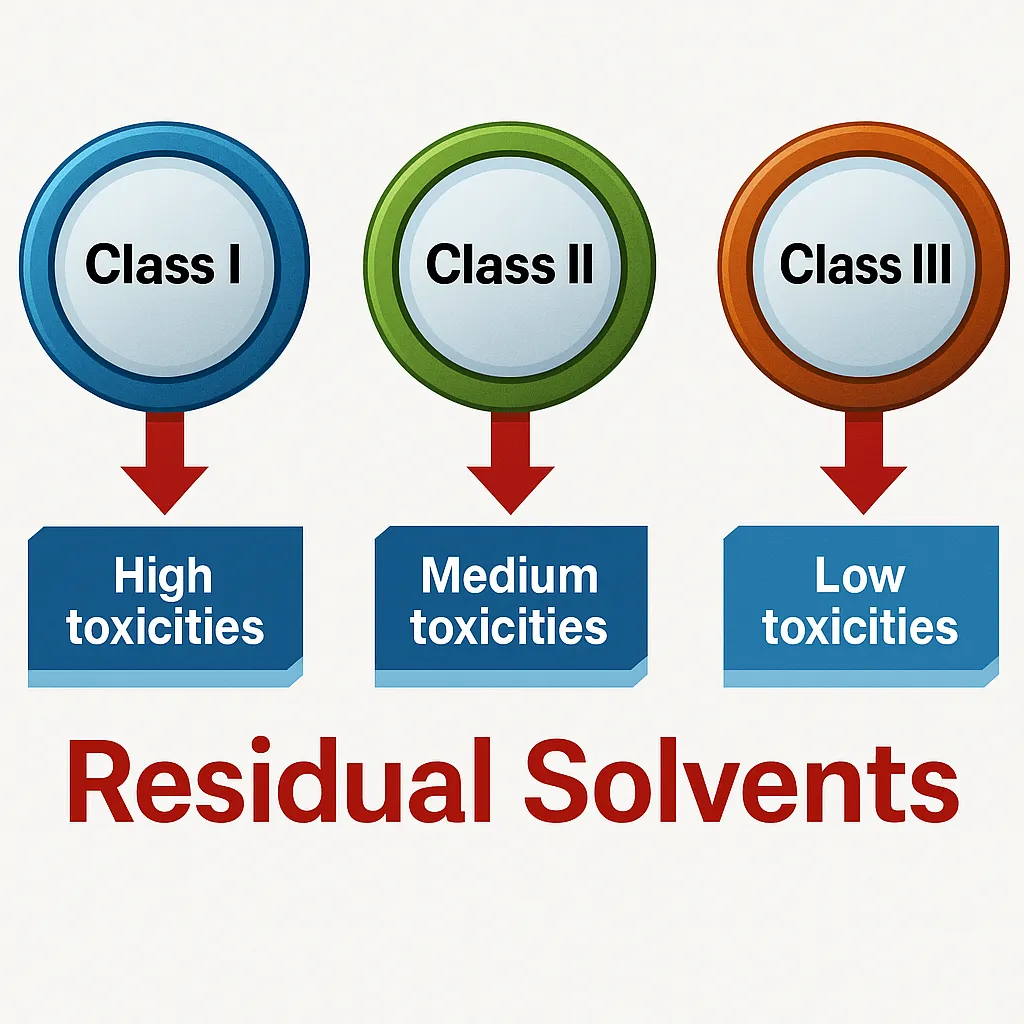
- Residual Solvents: These are solvents used during the API manufacturing or in the formulation of the drug product, that were not completely removed and therefore remain as impurities. Such solvents can be harmful if present in excess and are subject to regulatory limits and specifications based on their toxicity and potential health risks.
- Microbial Impurities: These contaminants may be introduced during the manufacturing process, either during API production or the formulation of finished products. They can also arise from improper handling, storage, or packaging. If not properly controlled, microbial contamination can lead to infections in patients or result in product spoilage.
- Heavy Metals: Small traces of metals such as lead, cadmium, mercury, etc, can be present due to contamination during the manufacturing process. Heavy metals are strictly regulated because of their high toxicity and potential to cause severe health issues.

- Packaging Materials Impurities: They arise from interactions between the drug product & its primary packaging. Such interaction may cause packaging materials to leach chemicals into the product, raising significant concerns about potential toxicity and risks to patients’ safety.
- Excipients Impurities: Low-quality excipients may pose a risk of introducing impurities, particularly if degradation or contamination occurs during storage.

Process-Related Impurities
We will talk in detail about each of the previously mentioned types, starting with the Process-Related ones.
As mentioned above, process-related impurities are substances that arise during the synthesis or purification of the Active Pharmaceutical Ingredient(API). To elaborate further, these impurities may originate from the following sources:
1-Chemical & Reagents
Reagents are defined as chemicals or substances that are added in the API synthesis to cause a chemical transformation, They include acids, bases, catalysts, solvents, etc
- Strong bases, like NaOH, and strong acids like HCl & H2SO4 are commonly used during the API synthesis process. If properly neutralized, these acids or bases may remain in the final product as a potential process-related impurity.

- Oxidizing agents like Hydrogen Peroxide or Reducing agents like NaBH₄-Sodium Borohydride- if found in trace amounts in the final product, API-, can lead to undesirable effects like the degradation of the API as well as raise toxicological concerns

- Solvents: Organic solvents are widely used in various stages of API manufacturing, including dissolution or extraction or purification, and crystallization. Common solvents include methanol, ethanol, chloroform, dimethylformamide (DMF), and dimethyl sulfoxide(DMSO). If these solvents are not efficiently removed through evaporation or drying processes, they may remain in the final product as Residual solvents. These residual solvents can pose serious health risks- for example, methanol can cause neurotoxicity, while acetone may lead to respiratory issues. Therefore, strict regulatory limits are set for residual solvent levels to ensure drug safety, following guidelines like ICH Q3C.
2-Catalysts:
Catalysts may include metal salts like copper sulfate or sodium chloride, as well as precious metals like platinum or palladium. These substances if present in the final product (API), can cause various side effects. Although typically used in small amounts due to their high cost, trace amounts may remain if purification was inadequate or if the reaction processes are poorly controlled. The presence of these metals poses a potential toxicological risk to the patients’ health.
")
3-Ligands & Additives:
Ligands are molecules that bind to metal centers, while additives-such as stabilizers and antioxidants, are used to enhance reaction conditions or product stability. If Ligands and additives used during chemical synthesis are not completely removed during the purification process, they are considered process-related impurities

4-Residual Reagents:
These reagents can cause irritation, toxicity, and other adverse effects when the product is administered. Residual reagents are unwanted chemicals that remain in the final product if they are not fully consumed during API synthesis.
Example: Sulfuric acid may be used as a catalyst during API synthesis. However, if proper purification or neutralization is not performed, traces of sulfuric acid may remain in the final product, posing a safety hazard.
5-Inorganic Impurities:
Stainless steel equipment-such as steel reactors and pipes, used during API manufacturing can introduce inorganic impurities, including metals like iron, aluminum, and lead. These impurities must be carefully monitored and controlled, as they can be highly toxic and may accumulate in the body over time.

6-Cross-Contamination:
When API production is performed on a large scale, cross-contamination is highly likely to occur. Ineffective cleaning procedures between different API batches can lead to traces of the previous active ingredient remaining in the current batch. Such cross-contamination results in the unintended presence of another active drug, which can cause adverse effects ranging from hypersensitivity reactions to more severe health risks.
7-By-products of chemical reactions:
Examples of by-products include Nonactive isomers- molecules that are chemically very similar to the intended API but lacking pharmacological activity well as over-oxidized intermediates or other by-products that are structurally different than the API.
If these by-products are not properly removed, they can accumulate during the production process, contaminating the final product, reducing its efficacy, and posing safety risks. Therefore, by-product impurities are carefully monitored and controlled throughout the API manufacturing.
8-By-products from Purification:
As previously mentioned, solvents, intermediate products, or reagents that are not completely removed during the manufacturing process may remain in the final product. These substances are then classified as impurities, potentially affecting the API’s efficacy and posing risks of adverse reactions to patient health.
Want to ensure the purity of your pharmaceutical substances? You can discover “Sources and Types of Impurities in Pharmaceutical Substances.“
Impact of Process-Related Impurities on the API
1-Toxicity:
Heavy metals, solvents, reagents, and other Impurities mentioned above pose significant risks to patient health, even when present in trace amounts.
2-Reduced Efficacy:
Impurities such as degradation products or by-products may interfere with API efficacy by altering the API’s structure, binding to the drug molecule, or even competing for the same biological targets.
3-Stability Issues:
The presence of process-related impurities can reduce the product’s shelf life, as they may accelerate API degradation through mechanisms such as oxidation, hydrolysis, and others.
Managing and Controlling Process-Related Impurities
1-Purification and Filtration:
Residual reagents and by-products could be effectively removed using various purification techniques such as distillation, recrystallization, and chromatography.
For Solvent Removal, several methods are employed, including vacuum distillation, rotary evaporation, and freeze-drying. These techniques are essential for eliminating solvents or other volatile substances from the final product, ensuring its purity and safety.

2-Optimization of reaction conditions:
Reaction parameters for API synthesis- including temperature, pressure, and reaction time- should be tightly controlled to minimize the formation of unwanted by-products and to ensure complete consumption of the reagents used.
3-Quality control tests:
The Identification and quantification of by-products and residual reagents in the final product is performed using various Analytical methods, including HPLC, GC, NMR, etc
High Performance Liquid chromatography (HPLC): A Comprehensive guide

4-Regulatory Compliance:
ICH guidelines concerned with the control of Impurities, specifically ICH Q3A-Impurities in new drug substances and ICH Q3B-Impurities in new drug products, should be adhered to. This is very important to ensure the safety and efficacy of the drug and compliance with regulatory standards.
Process Related Impurities regulatory limits & specifications

Process-related impurity limits are determined based on several factors, including pharmacological considerations, safety studies, and toxicological risk assessments.
Regulatory authorities such as the FDA, EMA & ICH provide detailed guidelines on Impurity testing and acceptable limits.
According to ICH Q3A, specifications for any individual impurity should not exceed 0.1% of the drug’s total content, while the limit for total Impurities should not exceed 1%.
A more detailed description of Impurity specifications and regulatory requirements from each authority will be discussed in the following sections.
Conclusion
Understanding the factors associated with impurities is fundamental to ensure the quality, safety, and efficacy of pharmaceutical products. Impurities originating from reagents, catalysts, solvents, equipment, and chemical processes during API synthesis can significantly impact drug stability, therapeutic effectiveness, and patient safety-even in minute amounts. Implementing effective strategies for controlling, identifying, and eliminating these impurities is essential-not only for minimizing toxicity and preventing degradation but also for complying with the strict requirements outlined in ICH guidelines ICH Q3A and Q3B.
In the following sections, we will discuss additional types of Impurities, with an in-depth explanation of Impurity specifications and how to calculate the percentage of Impurities in the finished drug product