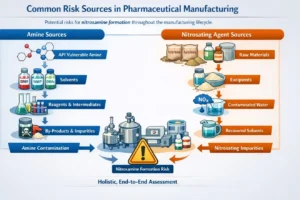
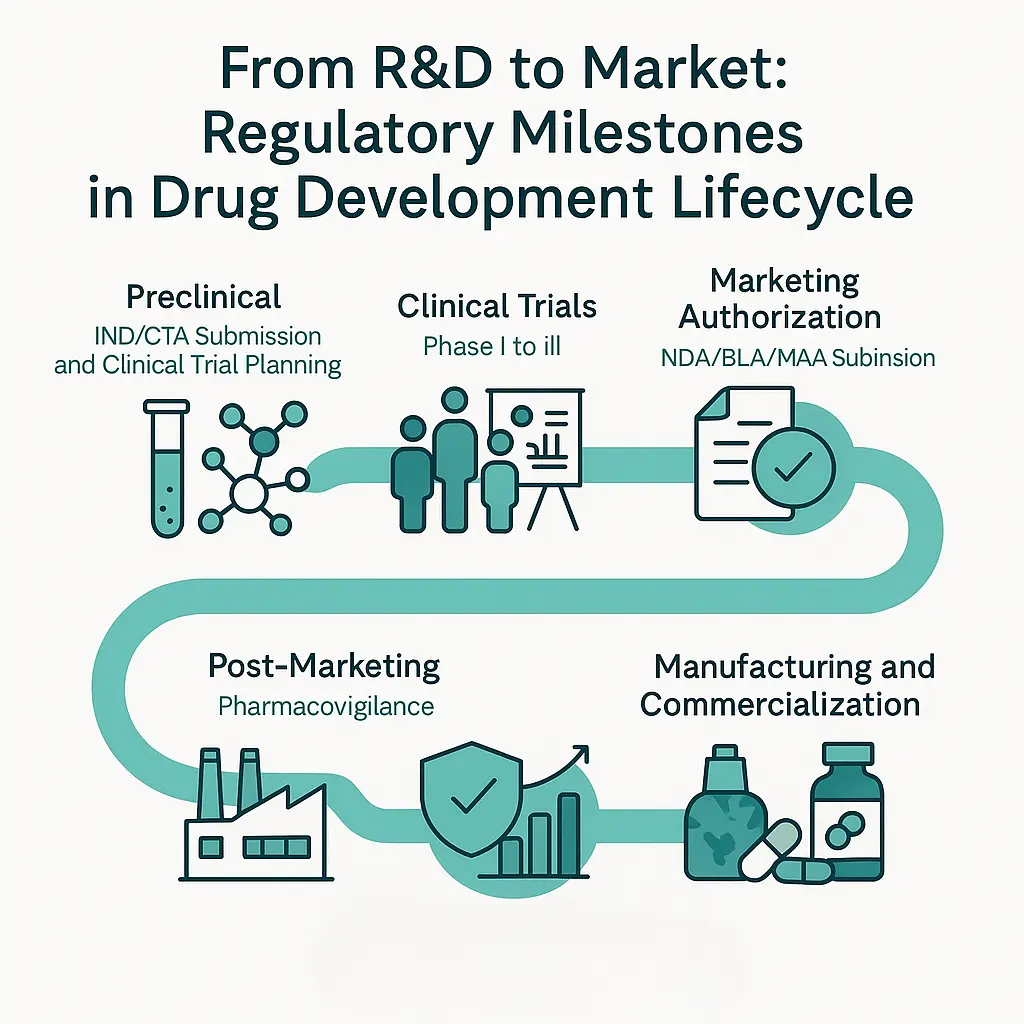
Pharmaceutical Regulatory Affairs (PRA):
Key Insights:
Pharmaceutical Regulatory Affairs (PRA) is a key field that guarantees the safety, effectiveness, and quality of medicines throughout their lifecycle. It connects drug manufacturers, regulatory agencies, and the public by managing the complex rules for drug approval, compliance, and monitoring after launch. PRA (Pharmaceutical Regulatory Affairs) experts play a vital role in maintaining global health and safety by making sure medicines meet strict regulatory standards before they reach patients.
Functions of Pharmaceutical Regulatory Affairs
The primary functions of Regulatory Affairs span the entire lifecycle of a pharmaceutical product, from development to post-marketing. Some of the critical functions include:
- Regulatory Submissions and Approvals
PRA professionals prepare and submit various regulatory dossiers, such as Investigational New Drug (IND) applications, New Drug Applications (NDA), Abbreviated New Drug Applications (ANDA), and Biologics License Applications (BLA). These submissions are tailored to meet country-specific regulatory guidelines.
- Compliance with Global Standards
Regulatory Affairs ensures compliance with Good Manufacturing Practices (GMP), Good Clinical Practices (GCP), and Good Laboratory Practices (GLP) as outlined by international bodies like the World Health Organization (WHO), International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH), and local regulatory bodies such as the US FDA and EMA.
- Product Labeling and Advertising
An essential function of Regulatory Affairs is to ensure that labeling, packaging, and marketing materials comply with regulatory standards. Misbranding or false advertising can lead to severe penalties and product recalls.
- Post-Marketing Surveillance and Pharmacovigilance
After a drug is approved and marketed, PRA professionals monitor its safety profile, report adverse drug reactions (ADRs), and implement necessary updates to its risk management plan (RMP).
Classification of Pharmaceutical Regulatory Affairs
Pharmaceutical Regulatory Affairs is a broad and multifaceted field that can be classified based on the stage of the product lifecycle, the type of pharmaceutical product, the regulatory focus area, or the geographic/regional regulatory jurisdiction. Below are the common ways PRA is classified:
1. Classification Based on Product Lifecycle Stages
- Research and Development (R&D) Regulatory Affairs:
Focuses on regulatory requirements during drug discovery and early development, including preclinical studies, Investigational New Drug (IND) applications, and clinical trial approvals.
- Clinical Regulatory Affairs:
Involves preparing submissions for clinical trial protocols, ethics committee approvals, ongoing clinical trial monitoring, and reporting serious adverse events (SAEs).
- Registration and Marketing Authorization:
Concerned with the preparation and submission of dossiers (e.g., NDA, MAA) to regulatory agencies to obtain product approval for marketing.
- Post-Marketing Regulatory Affairs:
Includes post-approval activities such as pharmacovigilance, safety updates, variation submissions (changes in labeling, manufacturing), renewals, and compliance inspections.
2. Classification Based on Type of Pharmaceutical Product
Regulatory requirements differ based on the category of pharmaceutical product:
- Small Molecule Drugs:
Conventional chemically synthesized drugs with established regulatory pathways.

- Biologics and Biopharmaceuticals:
Includes vaccines, monoclonal antibodies, gene therapies, and other complex biologic products requiring specialized regulatory considerations.

- Medical Devices and Combination Products:
Devices used for diagnosis or treatment, sometimes combined with drugs, require a different regulatory approach.
- OTC (Over the Counter) vs. Prescription Drugs:
OTC products must meet specific safety and labeling standards, while prescription medicines are subject to stricter controls.- medication labels: Active ingredient-purpose-intendeduse-Warning-Direction-strenght-inactive ingredients
- Nutraceuticals and Dietary Supplements:
Regulated differently in various countries, often with less stringent requirements than pharmaceuticals.

3. Classification Based on Regulatory Focus Areas
- Safety and Pharmacovigilance:
Focuses on monitoring, evaluating, and reporting adverse drug reactions (ADRs) and maintaining drug safety throughout the product lifecycle.
Immediate Action Required
All Market Authorization Holders (MAHs) of Montelukast—both local and foreign manufacturers—must:
- Implement the updated PIL with a box warning on all oral Montelukast dosage forms.
- Apply the update immediately, even for stocks currently available in pharmacies.
- Submit the amended PIL to NMRA within three months of the notification date.
Box Warning: Risk of Neuropsychiatric Reactions❗
Healthcare professionals must remain vigilant for the following adverse effects associated with Montelukast:
- Behavior changes
- Speech impairment
- Depression
- Obsessive-compulsive symptoms
These reactions may occur in adults, adolescents, and children taking Montelukast. If any of these symptoms present, evaluate the risks and benefits of continuing treatment.
Information for Patients and Caregivers
What should patients and parents/caregivers do?
If you or your child experiences any behavior or mood-related changes while taking Montelukast:
- Stop using the medication immediately
- Consult your healthcare provider without delay
Patients are strongly advised to read the updated PIL carefully, especially the list of neuropsychiatric reactions, and report any unusual symptoms to their physician.
For full details and official notification, visit the NMRA official website
- Quality Regulatory Affairs:
Deals with compliance regarding drug manufacturing practices, quality control, stability studies, and validation, ensuring Good Manufacturing Practices (GMP).
- Labeling and Promotional Regulatory Affairs:
Ensures that product labeling, packaging, and promotional materials comply with regulatory standards to avoid misleading claims. Each country has specific labelling requirements, and every health authority ensures the implementation of the guidelines.
Mandatory Label Element
- Brand Name
- Printed and prominently displayed.
- Generic Name with Pharmacopoeial Standard
- E.g., Montelukast Sodium USP
- Strength and Dosage Form
- Example: 10 mg Tablets
- Colorants
- Any colorants used in the formulation must be explicitly stated.
- Lactose Advisory
- If the product contains lactose, a “contains lactose” statement is required.
- Instructions for Use
- Including usual dosage and method of administration, if applicable.
- Storage Conditions
- Specify temperature and handling requirements.
- Manufacturer Information
- Manufacturer’s Name
- License Number
- Complete Address
- Country of Origin
- Bar Code / 2D Data Matrix
- Must be included on the label for product traceability.
Additional Notes:
- Ensure legibility and language compliance as required by NMRA.
- Labels must be approved during product registration and updated upon any change.

- Regulatory Intelligence:
Involves monitoring, analyzing, and interpreting current and upcoming regulatory changes globally to guide strategic planning.
- Compliance and Auditing:
Ensures ongoing compliance with regulatory requirements through internal audits and prepares for regulatory inspections.- The Pharmaceutical Inspection Co-operation Scheme (PIC/S) is a global initiative involving 54 member countries that work together to harmonize and uphold high-quality standards in pharmaceutical inspections.
- Inspections in these countries focus on ensuring Good Manufacturing Practices (GMP) compliance to guarantee medicine’s safety and quality.
- Because PIC/S promotes mutual recognition, inspections in member countries often follow a harmonized approach, reducing duplication and facilitating international pharmaceutical trade

4. Classification Based on Geographic or Jurisdictional Regulatory Frameworks
- US Regulatory Affairs: Compliance with FDA regulations, including the Code of Federal Regulations (CFR) Title 21 and other FDA guidance’s.
- European Regulatory Affairs: Focuses on EMA guidelines, European Medicines Agency centralized procedures, and national regulations within the European Union.
- Asia-Pacific Regulatory Affairs: Navigating regulations in countries like Japan (PMDA), China (NMPA), India (CDSCO), and others, each with unique requirements
- Global Regulatory Affairs: Harmonization efforts such as ICH (International Council for Harmonization) guidelines and coordination of multinational submissions.
Scope of Pharmaceutical Regulatory Affairs:
The scope of PRA has expanded significantly with the globalization of the pharmaceutical industry and advancements in technology. Its reach now extends beyond traditional drug development to include biologics, biosimilars, advanced therapy medicinal products (ATMPs), and digital therapeutics.
- Global Regulatory Harmonization
Organizations like the ICH, ASEAN, and PANDRH (Pan American Network for Drug Regulatory Harmonization) are working towards harmonizing regulatory requirements across regions. Regulatory professionals play a pivotal role in adapting submissions to meet these harmonized guidelines.
- Advanced Therapies and Biologics
Regulatory Affairs has evolved to address innovative therapies like gene therapy, monoclonal antibodies, and mRNA technologies. Each of these requires specialized regulatory pathways due to their unique mechanisms and manufacturing complexities.
- Emerging Markets / New Markets
Regulatory Affairs professionals are increasingly focusing on emerging markets, such as India, China, and Africa, which have rapidly grown pharmaceutical industries. These regions often have unique regulatory requirements that differ from established markets like the US and Europe.
- Digital Health and AI (Artificial Intelligence)
The rise of digital health solutions, including mobile health applications and AI-based diagnostics, poses new regulatory challenges. Regulatory Affairs professionals must navigate uncharted territories to ensure compliance with new and evolving frameworks.
Significance of Pharmaceutical Regulatory Affairs
Regulatory Affairs is vital not only for pharmaceutical companies but also for ensuring public health and safety through the following:
- Ensuring Patient Safety
By enforcing strict guidelines, Regulatory Affairs minimizes the risks associated with unsafe or ineffective medicines reaching the market.
- Facilitating Innovation
Regulatory Affairs professionals help companies bring innovative therapies to market by navigating complex approval pathways and ensuring compliance with regulations.
- Mitigating Legal and Financial Risks
Non-compliance with regulatory standards can result in hefty fines, product recalls, and reputational damage. PRA professionals mitigate these risks by ensuring adherence to all requirements.
- Enhancing Global Health
Through collaboration with international regulatory bodies, PRA professionals contribute to global public health initiatives, ensuring the availability of high-quality medicines worldwide.
Challenges in Pharmaceutical Regulatory Affairs
Regulatory Affairs professionals face numerous challenges, including:
Global Regulatory Diversity: Different countries have varying regulations and requirements, necessitating tailored regulatory strategies for each market, which can be complex and resource-intensive.
Rapidly Evolving / Changing Regulations: The regulatory landscape continuously changes in response to scientific advancement, public health emergencies, and policy shifts, requiring pharmaceutical regulatory affairs professionals to stay updated and agile.
Technological Advances: Emerging technologies such as personalized medicine, biologics, gene therapies, and digital health products introduce new regulatory considerations and uncertainties.
Balancing Speed and Safety: There is constant pressure to expedite drug approvals to meet patient needs while ensuring thorough evaluation of risks and benefits
Key International Regulatory Authorities
Pharmaceutical Regulatory Affairs operates in a global ecosystem governed by diverse regulatory authorities. Understanding these authorities is crucial for companies aiming to market their products internationally. Some of the most prominent agencies include:
- United States Food and Drug Administration (FDA)
Governs the approval and regulation of pharmaceuticals, biologics, and medical devices in the United States.
- European Medicines Agency (EMA)
Centralized body overseeing drug approvals within the European Union.
- Central Drugs Standard Control Organization (CDSCO)
The regulatory body in India responsible for approving drugs and clinical trials.
- Pharmaceuticals and Medical Devices Agency (PMDA)
Japan’s regulatory authority focuses on the safety and efficacy of drugs and medical devices.
- National Medical Products Administration (NMPA)
China’s regulatory agency, formerly known as the CFDA, oversees drug and device approvals.
- World Health Organization (WHO)
Provides global standards for drug quality and safety, aiding regulatory authorities worldwide.
References
- U.S. Food and Drug Administration (FDA). “Drug Approval Process.” www.fda.gov
- European Medicines Agency (EMA). “Guidelines and Regulatory Framework.” www.ema.europa.eu
- International Council for Harmonisation (ICH). “Harmonised Guidelines.” www.ich.org
- World Health Organization (WHO). “Good Manufacturing Practices.” www.who.int
- Central Drugs Standard Control Organization (CDSCO). “National Regulatory Framework.” www.cdsco.gov.in
- www.nmra.gov.lk/announcements/to-all-market-authorization-holders-of-montelukast
- https://www.nmra.gov.lk/pages/pharmacovigilance
Discover the Future of Mucosal Drug Delivery
Curious about how nanobodies and OMVs are transforming targeted therapies?
👉 Read the Full Article