
Introduction:
Millions of people worldwide suffer from the crippling hereditary condition known as sickle cell disease (SCD). SCD, which is characterized by red blood cells with a crescent shape, results in serious complications like stroke and organ damage, as well as ongoing pain and repeated hospital stays. Blood transfusions, bone marrow transplants, or symptomatic alleviation were the only available treatments for decades; these methods only offered short-term fixes or required a rare donor match.
CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats), a cutting-edge gene-editing technology that has redefined the boundaries of genetic medicine. CRISPR allows precise alterations in the DNA, enabling scientists to correct genetic mutations at their source. In the case of SCD, CRISPR has emerged as a beacon of hope, offering the potential for a one-time, curative therapy. This blog explores how CRISPR technology is transforming the landscape of personalized medicine for Sickle Cell Disease.
The Genetic Mutation Behind SCD:
SCD is caused by a single mutation in the HBB gene located on chromosome 11. This mutation results in the production of hemoglobin S instead of normal hemoglobin A. The abnormal hemoglobin causes red blood cells to assume a sickle shape, leading to reduced oxygen delivery, blood flow blockages, and destruction of red blood cells.
Traditional Treatments and Their Limitations:
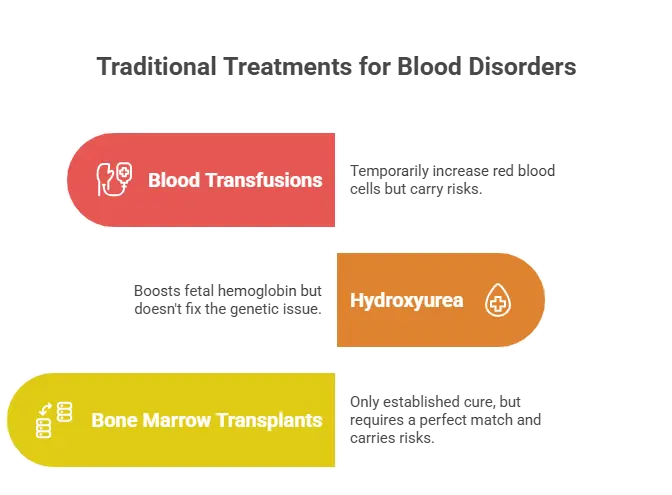
- Blood Transfusions: These temporarily increase the number of healthy red blood cells but carry risks like iron overload.
- Hydroxyurea: A drug that boosts fetal hemoglobin production but does not address the underlying genetic defect.
- Bone Marrow Transplants: The only established cure, yet it requires a perfectly matched donor and carries significant risks like graft-versus-host disease.
How CRISPR Works:
CRISPR functions as a “genetic scissor,” guided by a piece of RNA to the exact location of a mutation. Once there, it can:
- Cut and Replace: Remove the faulty DNA segment and replace it with a corrected sequence.
- Gene Activation or Suppression: Turn genes on or off to modify protein production.
- Base Editing: Correct a single nucleotide mutation without cutting the DNA strand entirely.
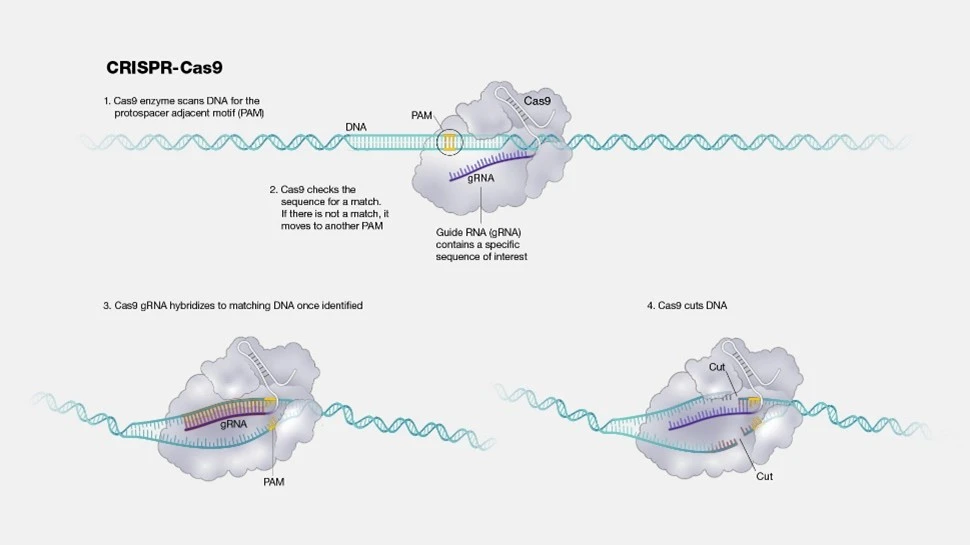
Fig.3. Illustration of how the CRISPR works.
CRISPR-Based Approaches to Treat SCD:
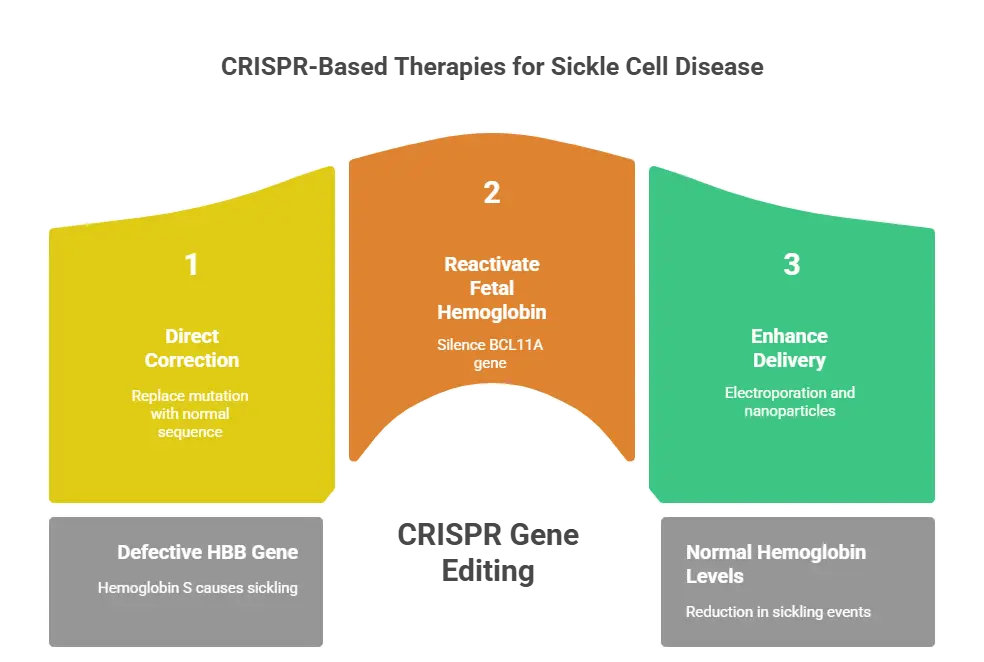
1. Direct Correction of the HBB Gene
Researchers use CRISPR to correct the defective HBB gene responsible for hemoglobin S. This approach involves:
Extracting the patient’s hematopoietic stem cells from their bone marrow or blood.
Editing the HBB gene using CRISPR to replace the mutation with a normal sequence.
Reinfusing the corrected stem cells back into the patient.
Clinical Success: Early-stage trials have shown that patients experience a significant increase in normal hemoglobin levels and a reduction in sickling events.
2. Reactivating Fetal Hemoglobin (HbF)
Another promising approach focuses on reactivating fetal hemoglobin (HbF), a form of hemoglobin naturally produced before birth. CRISPR targets the BCL11A gene, which suppresses HbF production after infancy. Silencing BCL11A allows the body to produce HbF, compensating for the lack of functional adult hemoglobin.
Clinical Success: Trials like CTX001 by CRISPR Therapeutics and Vertex Pharmaceuticals have demonstrated near-complete elimination of severe symptoms in treated patients.
3. Enhancing Delivery Methods
One of the challenges of CRISPR-based therapies is delivering the editing machinery to the correct cells efficiently. Researchers are developing:
Electroporation: A method to introduce CRISPR into stem cells by temporarily opening cell membranes.
Nanoparticle Delivery Systems: Using lipid nanoparticles to transport CRISPR components directly into the body.
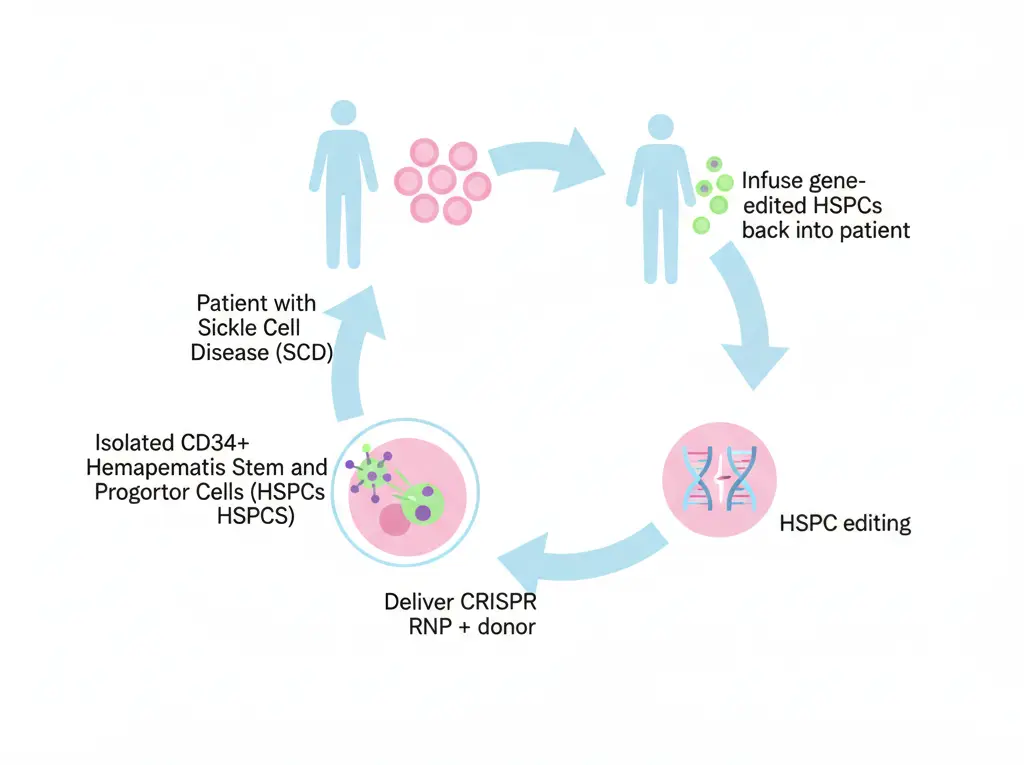
Fig.3. Genome editing-based strategy for treating sickle cell disease.
Despite its transformative potential, CRISPR technology raises important ethical and regulatory questions. Concerns include:
- Off-Target Effects: The possibility of unintended genetic modifications could have unforeseen consequences.
- Accessibility and Affordability: Ensuring that CRISPR-based therapies are accessible to populations most affected by SCD.
- Ethical Dilemmas: The broader implications of germline editing, which could affect future generations.
Conclusion:
CRISPR technology is transforming the treatment landscape for Sickle Cell Disease, offering patients a genuine chance at a cure. By addressing the root cause of the disease, CRISPR promises to eliminate suffering and restore hope for millions worldwide. As science progresses, this revolutionary tool will undoubtedly pave the way for a brighter future in personalized medicine.
References:
Demirci, S., Leonard, A., Haro-Mora, J. J., Uchida, N., & Tisdale, J. F. (2019). CRISPR/CAS9 for sickle cell disease: applications, future possibilities, and challenges. Advances in Experimental Medicine and Biology, 37–52. https://doi.org/10.1007/5584_2018_331
Park, S. H., & Bao, G. (2021). CRISPR/Cas9 gene editing for curing sickle cell disease. Transfusion and Apheresis Science, 60(1), 103060. https://doi.org/10.1016/j.transci.2021.103060
Rees, D. C., Williams, T. N., & Gladwin, M. T. (2010). Sickle-cell disease. The Lancet, 376(9757), 2018–2031. https://doi.org/10.1016/s0140-6736(10)61029-x
Sharma, A., Boelens, J., Cancio, M., Hankins, J. S., Bhad, P., Azizy, M., Lewandowski, A., Zhao, X., Chitnis, S., Peddinti, R., Zheng, Y., Kapoor, N., Ciceri, F., Maclachlan, T., Yang, Y., Liu, Y., Yuan, J., Naumann, U., Yu, V. W., . . . LaBelle, J. L. (2023). CRISPR-CAS9 editing of the HBG1 and HBG2 promoters to treat sickle cell disease. New England Journal of Medicine, 389(9), 820–832. https://doi.org/10.1056/nejmoa2215643 Xu, Y., & Li, Z. (2020). CRISPR-Cas systems: Overview, innovations and applications in human disease research and gene therapy. Computational and Structural Biotechnology Journal, 18, 2401–2415. https://doi.org/10.1016/j.csbj.2020.08.031

