Overview of Bioequivalence Guidance for Immediate-Release Solid Oral Dosage Forms (M13A)
Bioequivalence (BE) M13A guidance for immediate-release solid oral dosage forms, such as tablets and capsules, has been developed by the International Council for Harmonization (ICH). This guidance is adopted and implemented by regulatory agencies in ICH member countries, including the U.S. Food and Drug Administration (FDA). The M13A guidance provides a framework for conducting BE studies aimed at ensuring that generic and reference (innovator) products exhibit comparable bioavailability in terms of the rate and extent of drug absorption. It primarily focuses on pharmacokinetic (PK) endpoint studies.
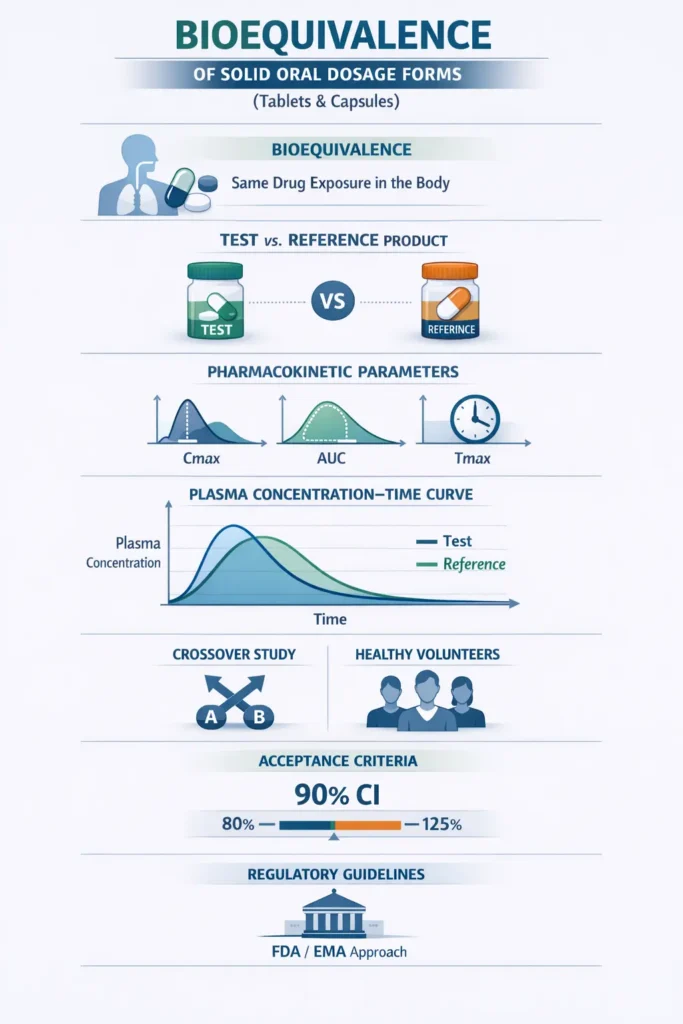
Objective of the M13A Bioequivalence Guidance
The primary objective of the M13A guidance is to offer recommendations for conducting BE studies for immediate-release solid oral drugs. These studies evaluate whether the test (generic) and reference (innovator) products demonstrate similar bioavailability. Specifically, the guidance emphasizes pharmacokinetic studies, which assess the rate and extent of absorption of a drug to establish bioequivalence.
Importance of Bioequivalence Assessment for Oral Dosage Forms
Bioequivalence assessments are essential for establishing therapeutic equivalence between generic and reference products. This ensures that a generic product will perform the same as the innovator product in terms of safety, efficacy, and overall therapeutic effect.
BE assessments are also important for innovator drugs in situations such as:
- To confirm bioavailability when comparing different formulations of the same drug.
For example, if marketing requests a capsule dosage form instead of the tablet formulation used in clinical studies, a bioequivalence (BE) study would typically suffice. This approach eliminates the need to repeat costly clinical trials on the capsule formulation, as BE studies require significantly fewer subjects and are much more cost-effective. - To confirm that modifications in formulation or manufacturing processes do not alter the product\’s bioavailability
General Principles for Establishing Bioequivalence
The M13A guidance outlines several general principles for establishing bioequivalence, which are crucial for designing and conducting BE studies.

Study Population
Bioequivalence studies should ideally be conducted in healthy subjects to minimize variability and accurately detect differences between drug formulations. However, if a drug poses safety concerns or unacceptable risks for healthy individuals, the study may be conducted in a targeted patient population under appropriate medical supervision.
Even healthy subjects must meet the inclusion and exclusion criteria outlined in the study protocol. These criteria should be clearly defined within the protocol. Subjects should be at least 18 years old and ideally have a Body Mass Index (BMI) between 18.5 and 30.0 kg/m². If the drug product is intended for use by both sexes, the study design should consider including both male and female participants.
Study Design
In bioequivalence (BE) studies, a randomized, single-dose, crossover design is recommended to compare the test (new) and comparator (reference) formulations. Each subject receives both formulations in two separate periods, with a washout phase in between. This design minimizes variability by allowing subjects to act as their controls, requires fewer participants, and provides reliable pharmacokinetic data (Cmax, Tmax, AUC) for assessing bioequivalence. It is efficient, cost-effective, and widely accepted by regulatory agencies.
Single-dose studies are typically the most sensitive method for detecting differences in the rate and extent of absorption between the test and reference products. The study design should include the following key considerations:
- Washout Period: A sufficient washout period (usually at least five elimination half-lives) is required between treatment periods to ensure that the drug from the first treatment is cleared from the body before the second dose is administered.
- Long Half-Life Drugs: For drugs with long elimination half-lives, a randomized parallel design may be used instead of a crossover design to avoid long washout periods.
Sample Size
The minimum number of evaluable subjects in a BE study depends on the study design:
- For a crossover design, at least 12 subjects should be included.
- For a parallel design, 12 subjects per treatment group are required.
Test and Comparator Products
The comparator product is the reference drug that is accepted by regulatory agencies and used as a benchmark for comparing the test product in the BE study. The test product should represent the formulation that is intended for marketing.
Fasting and Fed Study Conditions
For most immediate-release (IR) solid dosage forms, fasting conditions are preferred, as they provide better discrimination between the test and comparator products\’ pharmacokinetic profiles. However, different study conditions may be required based on the nature of the drug:
Non-High-Risk Products:
- If the drug is labeled for use under fasting conditions or with no food restrictions, a single BE study under fasting conditions is usually sufficient.
- If the drug must be taken with food for pharmacokinetic reasons (e.g., to enhance absorption or reduce variability), a BE study under fed conditions is recommended.
- If food is required for tolerability reasons (e.g., to prevent stomach irritation), either fasting or fed conditions are acceptable, depending on the drug\’s labeling.
High-Risk Products:
- High-risk products, such as drugs with low solubility or complex formulations (e.g., lipid-based or nanotechnology products), may perform differently under fasting and fed conditions. In such cases, studies under both conditions are required, as results from one condition cannot predict those of the other.
- It is acceptable to conduct either two separate two-way crossover BE studies or
- One four-way crossover BE study to evaluate both fasting and fed conditions.
Dose and Strength to be Studied
The highest strength of the product to be marketed should be used in the BE study, unless the highest strength is deemed unsafe. If a lower strength is used, dose proportionality must be demonstrated.
Dose proportionality means that the dose-adjusted Cmax (maximum concentration) and AUC (area under the curve) should differ by no more than 25% across different strengths of the product.
- Non-Proportional Pharmacokinetics (PK):
- Greater than proportional PK: Test at the highest strength.
- Less than proportional PK:
– If due to solubility issues or unknown reasons, test both the lowest and highest strengths.
– If due to absorption saturation, test at the lowest strength.
Moieties to be Measured
- Primary Approach – Parent Drug: BE is typically demonstrated using the parent drug since its concentration-time profile is more sensitive for detecting formulation differences than its metabolites.
- Active Enantiomers: If one enantiomer is inactive or contributes minimally to safety and efficacy, BE can be demonstrated for the active enantiomer only.
Standardization in BE Studies
To ensure consistency and reliable results, BE studies must be standardized in terms of conditions, dosing, and meal timing.
- Fasting Conditions:
- Subjects should fast for at least 8 hours before dosing.
- Water is allowed freely except for 1 hour before and after dosing. The dose should be taken with 150-250 mL of water.
- No food is allowed for 4 hours after dosing, and subsequent meals must be standardized.
- Fed Conditions:
- Subjects should consume a standardized meal 30 minutes before dosing:
- For high-risk products, a high-fat, high-calorie meal (900-1000 kcal, with 50% fat) is recommended.
- For non-high-risk products, a low-fat, low-calorie meal (approximately 500 kcal) is used, or the meal may follow the comparator product\’s labeling instructions.
- Subjects should consume a standardized meal 30 minutes before dosing:
Bioequivalence Criteria
The bioequivalence criterion for immediate-release solid oral dosage forms is typically set within an 80%-125% range for both the rate (Cmax) and extent (AUC) of drug absorption.
Conclusion
The M13A Bioequivalence guidance provides a comprehensive framework for evaluating the bioequivalence of immediate-release solid oral dosage forms. By focusing on pharmacokinetic studies and providing detailed recommendations on study design, sample size, fasting and fed conditions, and dosing requirements, the guidance ensures that both generic and innovator products meet stringent standards for bioavailability. This helps guarantee that patients receive safe, effective, and therapeutically equivalent treatments.
For FDA Reference: M13A Bioequivalence for Immediate-Release Solid Oral Dosage Form

