Pharmacokinetics (PK), the study of how the body affects a drug, is a cornerstone of drug discovery and development. It encompasses the processes of Absorption, Distribution, Metabolism, and Excretion (ADME), which collectively determine a drug’s concentration at its site of action and its therapeutic efficacy and safety. With the increasing complexity of novel compounds, understanding their unique pharmacokinetic profiles has become more critical than ever.
This article provides a comprehensive overview of fundamental pharmacokinetics (PK) principles, highlights the challenges and considerations in assessing novel compounds, and explores advanced modeling and prediction techniques, including Physiologically Based Pharmacokinetic (PBPK) modeling and the transformative role of Artificial Intelligence (AI) and Machine Learning (ML). By integrating these cutting-edge approaches, the pharmaceutical industry can accelerate the development of safer and more effective therapies, ultimately benefiting patient care.
Introduction
The journey of a drug from its administration to its elimination from the body is a complex and dynamic process. Pharmacokinetics (PK) is the scientific discipline dedicated to understanding this journey, quantifying how the body absorbs, distributes, metabolizes, and eliminates a drug over time. It is often summarized by the acronym ADME: Absorption, Distribution, Metabolism, and Excretion. These four fundamental processes dictate the concentration of a drug at its target site, influencing its therapeutic efficacy, potential toxicity, and overall duration of action.
A thorough understanding of PK is therefore indispensable at every stage of drug development, from early discovery to clinical application. These new therapeutic modalities often present unprecedented pharmacokinetic challenges, necessitating sophisticated approaches to characterize their behavior within biological systems. We will first revisit the fundamental ADME processes, laying the groundwork for understanding drug disposition, highlighting the critical role of early PK evaluation in drug development.
Fundamentals of Pharmacokinetics: ADME Processes
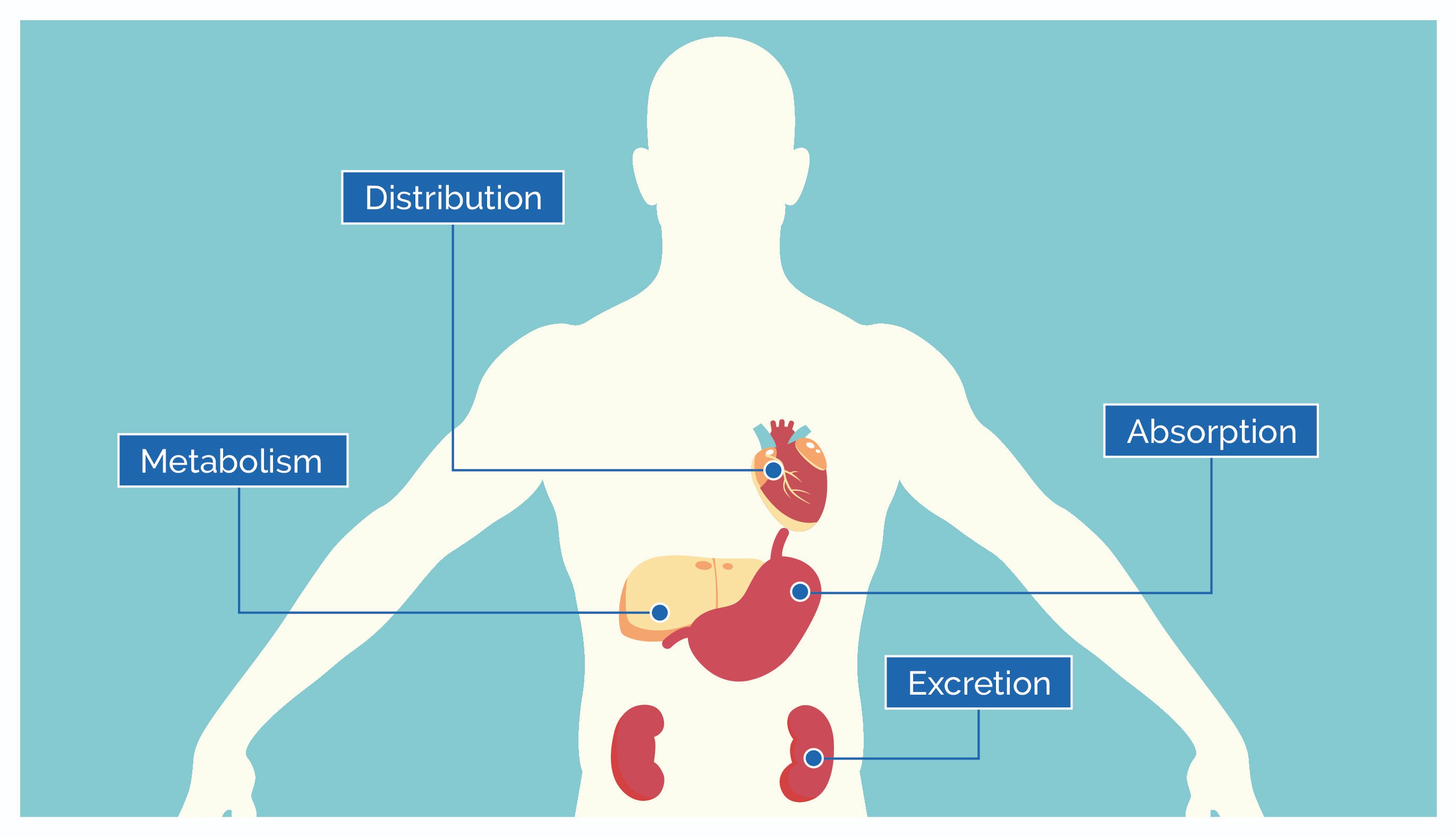
Pharmacokinetics describes the quantitative analysis of how a drug moves through the body. Read More. This dynamic process is conventionally broken down into four key stages: Absorption, Distribution, Metabolism, and Excretion (ADME). Each stage plays a critical role in determining the drug’s concentration at its site of action and its overall therapeutic profile.
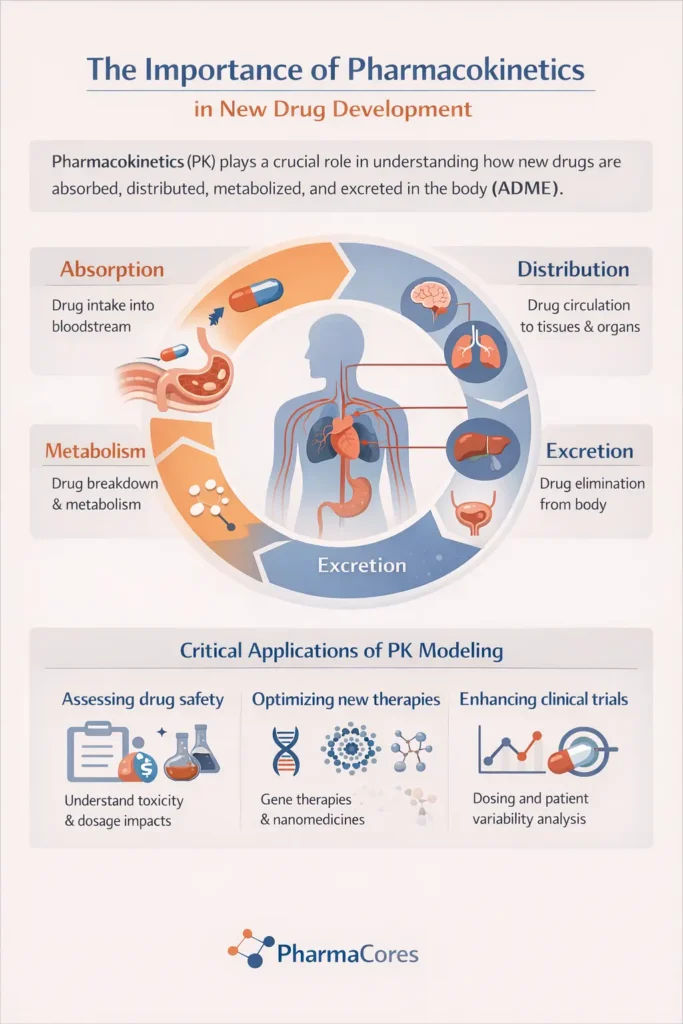
Absorption
Absorption is the process by which a drug moves from its site of administration into the systemic circulation. The rate and extent of absorption are crucial, as they dictate how quickly and how much of the drug reaches its intended target. Various routes of administration, such as oral, intravenous, intramuscular, subcutaneous, and transdermal, each possess distinct absorption characteristics, advantages, and disadvantages. For instance, intravenous administration bypasses the absorption phase entirely, leading to immediate and complete systemic availability.
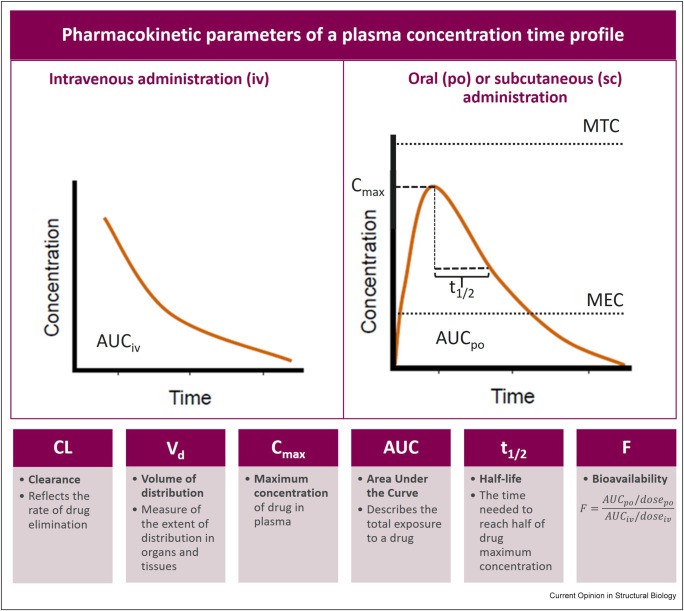
For orally administered drugs, absorption is a complex process involving liberation from the dosage form, dissolution, and passage across biological membranes, primarily in the gastrointestinal tract. Factors such as gastric pH, gut motility, food presence, and the drug’s physicochemical properties (e.g., solubility, lipophilicity, molecular size) significantly influence oral absorption. Bioavailability, defined as the fraction of the administered dose that reaches the systemic circulation unchanged, is a key metric reflecting the extent of absorption. Oral drugs often undergo ‘first-pass metabolism’ in the liver or gut wall, which can substantially reduce their bioavailability before reaching systemic circulation.
Distribution
Once absorbed, a drug is distributed throughout the body via the bloodstream to various tissues and organs. The extent and pattern of distribution are influenced by the drug’s physicochemical properties, such as its lipophilicity, molecular size, and ability to bind to plasma proteins or tissue components. The volume of distribution (Vd) is a theoretical pharmacokinetic parameter that relates the amount of drug in the body to the concentration of drug in the blood or plasma. A high Vd indicates extensive distribution into tissues, while a low Vd suggests the drug remains largely in the bloodstream.
Protein binding is a significant factor in drug distribution. Many drugs bind reversibly to plasma proteins, primarily albumin and alpha-1-acid glycoprotein. Only the unbound, or ‘free,’ fraction of the drug is pharmacologically active, capable of interacting with receptors, crossing biological membranes, and being eliminated. Changes in protein binding due to disease states (e.g., renal or hepatic impairment), age, or drug-drug interactions can alter the free drug concentration, potentially leading to altered therapeutic effects or increased toxicity.
Metabolism
Metabolism, also known as biotransformation, is the process by which the body chemically modifies drugs, typically to facilitate their elimination. The liver is the primary site of drug metabolism, owing to its rich supply of metabolizing enzymes, particularly the cytochrome P450 (CYP450) enzyme system. Drug metabolism can lead to the formation of inactive metabolites, more water-soluble compounds for easier excretion, or, in some cases, active metabolites (prodrugs).
Drug metabolism is broadly categorized into two phases: Phase I reactions (e.g., oxidation, reduction, hydrolysis) introduce or expose polar functional groups, while Phase II reactions (e.g., glucuronidation, sulfation, acetylation) involve conjugation with endogenous substances to form more polar and readily excretable compounds. Genetic variations, age, disease, and concomitant drug use can significantly impact metabolic enzyme activity, leading to inter-individual variability in drug response and potential drug-drug interactions.
Excretion
Excretion is the final stage of pharmacokinetics, involving the irreversible removal of the drug and its metabolites from the body. The kidneys are the most important organs for drug excretion, primarily eliminating water-soluble compounds. Renal excretion involves three main processes: glomerular filtration, tubular reabsorption, and tubular secretion. Other routes of excretion include biliary (via bile into feces), pulmonary (for volatile compounds), and mammary (via breast milk).
The efficiency of renal excretion is influenced by factors such as renal blood flow, glomerular filtration rate, and urinary pH. Impaired renal function can lead to drug accumulation and increased risk of toxicity, necessitating dose adjustments. Hepatic excretion, particularly for drugs with high molecular weight or extensive protein binding, involves secretion into bile and subsequent elimination in the feces. The enterohepatic recirculation, where drugs or their metabolites are reabsorbed from the intestine after biliary excretion, can prolong their systemic exposure.
Pharmacokinetics in Novel Drug Development
The advent of novel compounds, including complex biologics, gene therapies, and highly potent small molecules, has revolutionized drug discovery. However, these innovations also present unique pharmacokinetic challenges that necessitate a refined approach to their assessment. Understanding the PK profile of novel compounds is paramount for ensuring their safety, efficacy, and successful translation from bench to bedside.
Challenges and Considerations for Novel Compounds
Novel compounds often exhibit distinct physicochemical properties and mechanisms of action that can significantly alter their ADME characteristics compared to traditional small-molecule drugs. For instance:
- Biologics (e.g., antibodies, proteins): These large molecules typically undergo proteolytic degradation rather than hepatic metabolism by CYP enzymes. Their distribution is often limited to the extracellular fluid, and their elimination can involve target-mediated drug disposition (TMDD), where the drug binds to and is internalized by its pharmacological target, leading to non-linear pharmacokinetics. Immunogenicity, the development of anti-drug antibodies, can also significantly impact their PK profile.
- Gene Therapies: The PK of gene therapies is highly complex, involving the delivery, expression, and persistence of genetic material. The vector used for delivery (e.g., adeno-associated virus, lentivirus) plays a crucial role in its distribution and clearance. Understanding the biodistribution of the vector and the expressed therapeutic protein, as well as potential immune responses, is critical.
- Nanomedicines: Drugs encapsulated in nanoparticles or other nanocarriers have altered PK properties due to their size, surface charge, and composition. They often exhibit prolonged circulation times, altered distribution patterns (e.g., enhanced permeability and retention effect in tumors), and unique elimination pathways.
- Highly Potent Small Molecules: While still small molecules, their high potency often means very low doses are administered, making accurate measurement of their concentrations challenging. They may also interact with a wider range of transporters or enzymes, leading to complex DDI profiles.
These complexities underscore the need for tailored PK assessment strategies for each novel compound class, moving beyond conventional approaches.
Role of Early PK Assessment
Integrating PK assessment early in the drug discovery and development process is crucial for mitigating risks and accelerating the progression of promising drug candidates. Early PK studies, often conducted in preclinical models, provide vital information that informs lead optimization, candidate selection, and dose prediction for first-in-human studies. Key aspects of early PK assessment include:
- Candidate Prioritization: PK data helps in selecting compounds with favorable ADME properties, reducing the likelihood of late-stage failures due to poor bioavailability, rapid clearance, or unfavorable distribution.
- Structure-Activity Relationship (SAR) and Structure-Property Relationship (SPR) Optimization: PK insights guide medicinal chemists in modifying chemical structures to improve ADME characteristics while maintaining pharmacological activity.
- Dose and Regimen Design: Early PK data from preclinical species, often combined with allometric scaling, provides initial estimates for human dosing, helping to design safe and effective clinical trial protocols.
Identification of Potential Drug-Drug Interactions: Early in vitro and in vivo studies can identify potential DDI liabilities, allowing for risk mitigation strategies or further investigation.
By proactively addressing PK considerations, drug developers can make informed decisions, optimize compound design, and streamline the drug development pipeline, ultimately bringing novel therapies to patients more efficiently.
Advanced Pharmacokinetic Modeling and Prediction
The increasing complexity of novel compounds and the drive for more efficient drug development have propelled the field of pharmacokinetics towards advanced modeling and prediction methodologies. These approaches leverage computational power and sophisticated algorithms to simulate, predict, and interpret drug behavior in biological systems, significantly enhancing our ability to make informed decisions throughout the drug development pipeline.
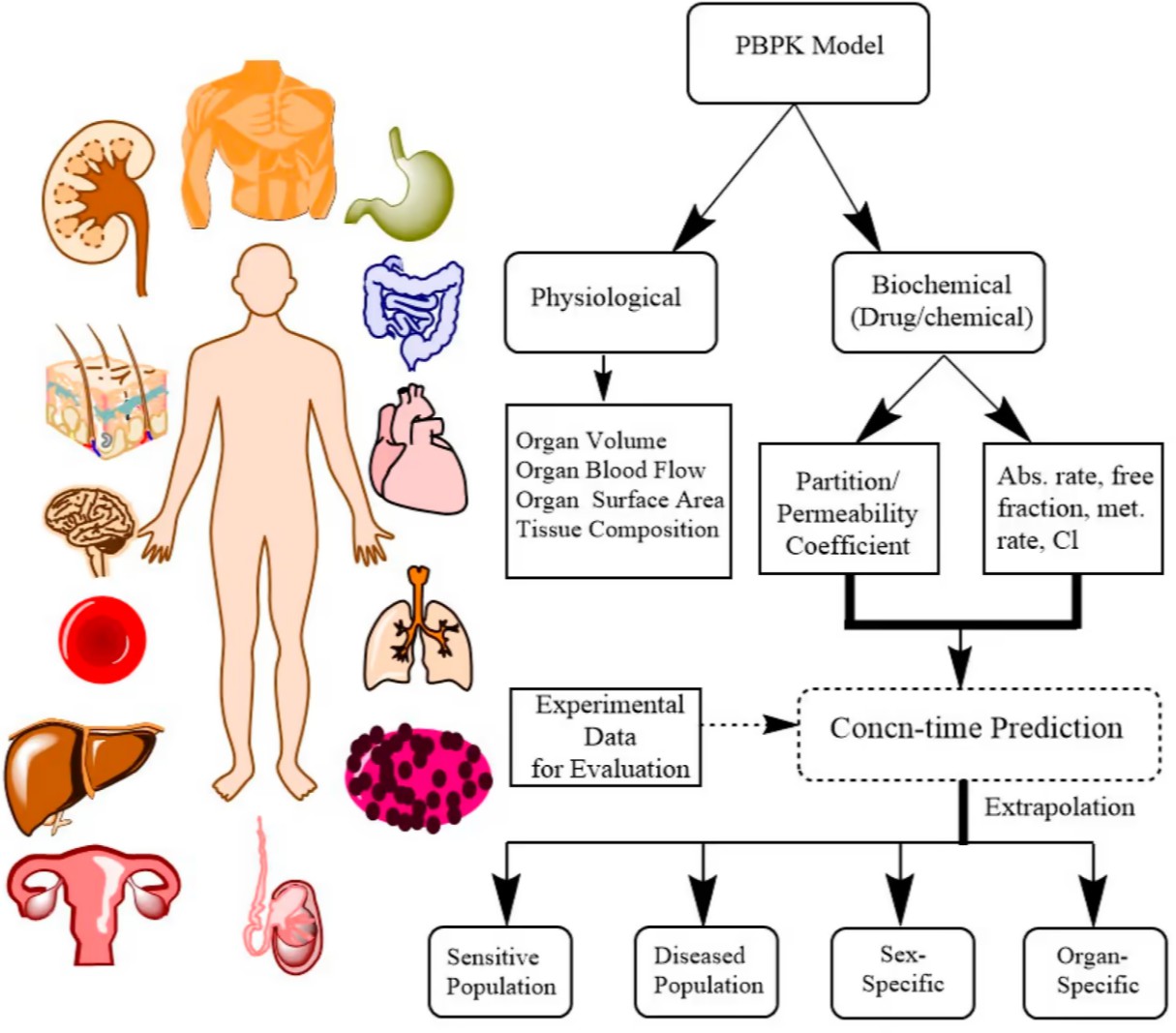
Physiologically Based Pharmacokinetic (PBPK) Modeling
Physiologically Based Pharmacokinetic (PBPK) modeling is a mechanistic, bottom-up approach that simulates the ADME processes within a physiologically realistic framework. Unlike traditional compartmental models, PBPK models integrate drug-specific parameters (e.g., physicochemical properties, enzyme kinetics, transporter affinities) with system-specific physiological parameters (e.g., organ volumes, blood flows, tissue composition). This allows for the prediction of drug concentrations in various tissues and organs over time, providing a more comprehensive understanding of drug disposition.
Key Advantages of PBPK Modeling:
- Extrapolation: PBPK models facilitate extrapolation across species (from animals to humans), populations (e.g., pediatrics, geriatrics, renally or hepatically impaired patients), and different dosing regimens.
- Drug-Drug Interaction (DDI) Prediction: PBPK models are highly effective in predicting and explaining DDIs, particularly those mediated by cytochrome P450 (CYP) enzymes and transporters. They can simulate the impact of perpetrator drugs on victim drug exposure, informing dosing adjustments and labeling recommendations.
- Formulation and Food Effect Prediction: PBPK models can predict the impact of different formulations or food intake on drug absorption and bioavailability.
- Reduced Clinical Trials: By providing robust predictions, PBPK modeling can reduce the need for certain clinical studies, thereby accelerating drug development and reducing costs.
PBPK modeling is increasingly accepted by regulatory agencies worldwide as a valuable tool for supporting drug development and regulatory decision-making.
Machine Learning and Artificial Intelligence in PK Prediction
The explosion of data in drug discovery and the advancements in computational power have paved the way for the application of Machine Learning (ML) and Artificial Intelligence (AI) in pharmacokinetic prediction. These data-driven approaches can identify complex patterns and relationships within large datasets, offering powerful tools for predicting PK properties and optimizing drug design.
Applications of AI/ML in PK:
- Early PK Property Prediction: ML models can predict ADME properties (e.g., solubility, permeability, metabolic stability, protein binding) directly from chemical structures, enabling early screening and prioritization of compounds with favorable PK profiles.
- PK Profile Prediction: Advanced ML algorithms can predict the entire concentration-time profile of a drug in vivo, reducing the reliance on extensive animal experimentation.
- Identification of PK Biomarkers: AI can help identify novel biomarkers that correlate with drug exposure and response, facilitating personalized medicine approaches.
- Optimization of Dosing Regimens: ML models can analyze patient-specific data to recommend optimized dosing regimens, minimizing adverse effects and maximizing therapeutic outcomes.
While still an evolving field, AI and ML hold immense promise for transforming pharmacokinetic research, enabling faster, more accurate, and more efficient drug development.
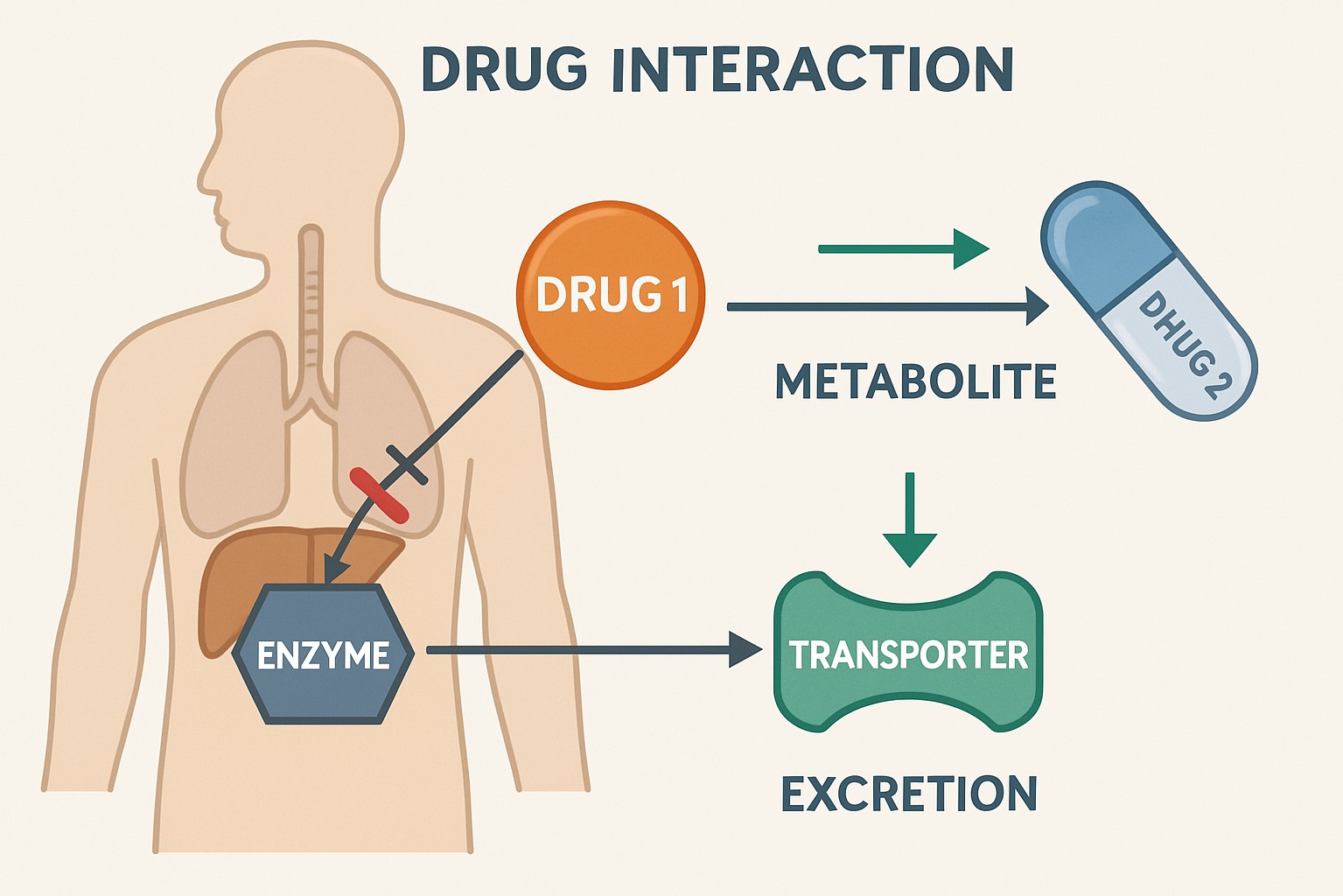
Drug-Drug Interactions (DDIs) and PK
Drug-drug interactions (DDIs) occur when the effects of one drug are altered by the presence of another. Pharmacokinetic DDIs are particularly significant, as they can lead to changes in drug exposure, potentially resulting in therapeutic failure or increased toxicity. These interactions often involve the modulation of drug-metabolizing enzymes (e.g., CYP450 enzymes) or drug transporters.
Understanding and predicting DDIs is a critical aspect of drug development and clinical practice. In vitro studies (e.g., enzyme inhibition/induction assays, transporter assays) are typically conducted early in development to assess DDI potential. These data, combined with PBPK modeling and clinical DDI studies, help to characterize the magnitude and clinical relevance of potential interactions. Regulatory guidelines emphasize the importance of DDI assessment to ensure patient safety and inform appropriate dosing recommendations and warnings in drug labeling.
Future Outlook and Conclusion
Pharmacokinetics remains a dynamic and evolving field, continually adapting to the complexities of novel compounds and the demands of modern drug development. The integration of advanced computational tools, particularly PBPK modeling and AI/ML, is transforming how we understand and predict drug behavior in biological systems. These technologies offer unprecedented opportunities to accelerate drug discovery, optimize therapeutic regimens, and enhance patient safety.
Looking ahead, the field of pharmacokinetics will likely see further advancements in several key areas:
- Integration of Multi-omics Data: Combining PK data with genomics, proteomics, and metabolomics will provide a more holistic understanding of drug response and inter-individual variability.
- Enhanced Predictive Capabilities: Continuous refinement of AI/ML algorithms and PBPK models, coupled with larger and more diverse datasets, will lead to even more accurate and robust predictions of drug disposition.
- Personalized Medicine: PK principles, guided by advanced modeling and patient-specific data, will play an increasingly central role in tailoring drug therapy to individual patients, maximizing efficacy while minimizing adverse effects.
- Regulatory Science: Regulatory agencies will continue to embrace and integrate advanced PK modeling and simulation into their assessment processes, facilitating the approval of innovative therapies.
In conclusion, pharmacokinetics is not merely a descriptive science but a predictive and integrative discipline essential for the rational design and development of new drugs. By embracing cutting-edge technologies and fostering interdisciplinary collaboration, the field of pharmacokinetics will continue to drive innovation, ultimately contributing to the delivery of safer, more effective, and personalized medicines to patients worldwide.
Key Takeaways
- Understanding pharmacokinetics is crucial for optimizing the dosage, efficacy, and safety of new compounds.
- Innovative analytical techniques enhance the characterization of drug absorption, distribution, metabolism, and excretion (ADME).
- Early pharmacokinetic profiling accelerates the drug development process by identifying potential issues sooner.
- Physiologically based pharmacokinetic (PBPK) modeling aids in predicting human responses from preclinical data.
- Integrating pharmacokinetic studies with pharmacodynamic insights improves the overall success rate of new drug candidates
References
- FDA. (2019). Impact Story: Supporting Drug Development Through Physiologically Based Pharmacokinetic Modeling. Available from: https://www.fda.gov/drugs/regulatory-science-action/impact-story-supporting-drug- development-through-physiologically -based-pharmacokinetic-modeling
- Grogan, S., & Preuss, C. V. (2023). Pharmacokinetics. In StatPearls. StatPearls Publishing. Available from: https://www.ncbi.nlm.nih.gov/books/NBK557744/
- High, K. A., & Roncarolo, M. G. (2019). Gene Therapy. The New England Journal of Medicine, 381(5), 455-4a4.
- Jamei, M., Marciniak, S., Adkin, D., Barter, Z., Daffertshofer, D., Dickinson, N., … & Rostami-Hodjegan, A. (2013). The Simcyp Population-Based ADME Simulator. Clinical Pharmacokinetics, 52(1), 1-1a.
- Pillai, N., Abos, A., Teutonico, D., & Mavroudis, P. D. (2024). A machine learning framework to predict the pharmacokinetic profile of small-molecule drugs based on chemical structure. Clinical and Translational Science. Available from: https://ascpt.onlinelibrary.wiley.com/doi/10.1111/cts.13824
- Singh, S., & Singh, A. (2023). Pharmacokinetics of Biologics. Journal of Pharmaceutical Sciences, 112(1), 1-15.
- U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER). (2020). Guidance for Industry: Drug-Drug Interaction Studies — Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations. Available from: https://www.fda.gov/media/134582/download
- Wicki, A., Witzigmann, D., Balasubramanian, V., & Huwyler, J. (2020). Nanomedicine in Cancer Therapy: Challenges and Opportunities. Advanced Drug Delivery Reviews, 154-155, 1-17.

